
Transcriptional noise, isoform prediction, and the utility of mass spec data in gene annotation
The human genome may be 80% functional or 8.2% functional. Maybe it's 93.7% functional or only 6.1%. I guess that all we know for sure is that it is not 0% functional (although my genome on a Monday morning may provide evidence to the contrary).
Transcript data can be used to ascribe some sort of functionality to a genome and, in an ideal world, we would sequence full-length cDNAs for every gene. But in the less-than-ideal world we often end up sequencing lots of small bits of RNA using an ever-changing set of technologies. ESTs, SAGE, CAGE, RACE, MPSS, and RNA-Seq have all been used to provide evidence for where genes are and how highly they are being expressed.
Having some transcriptional evidence is (usually) better than not having any transcriptional evidence, but it doesn't necessarily imply functionality. A protein-coding gene that is transcribed may not be translated. Transcript data is used in gene annotation to add new genes, especially in the case of a first-pass annotation of a new genome. But in established genomes, it is probably used more to annotate transcript isoforms (e.g. splice variants). This can lead to a problem for the end users of such data…how to tell if all isoforms are equally likely?
Consider the transcript data for the rpl-22 gene in Caenorhabditis elegans. This gene has two annotated splice variants and there is indeed EST evidence for both variants, but it is a little bit unbalanced:
This gene encodes the large ribosomal subunit protein…a pretty essential protein! Notice how the secondary isoform (shown on top) a) encodes for a much shorter protein and b) has very little transcript evidence. In my mind, this secondary isoform is the result of 'transcriptional noise'. Maybe a couple of lucky ESTs captured the transcript in the process of heading towards destruction via nonsense-mediated decay? It seems highly unlikely that this secondary transcript gives rise to a functional protein though someone who is new to viewing data like this might initially consider each isoform as equally valid.
If we turn on some additional data tracks to look at protein homology to human (shown in orange) and mass spectromety data from C. elegans (shown in red) it becomes clear that all of the evidence is really pointing towards just one functional isoform:
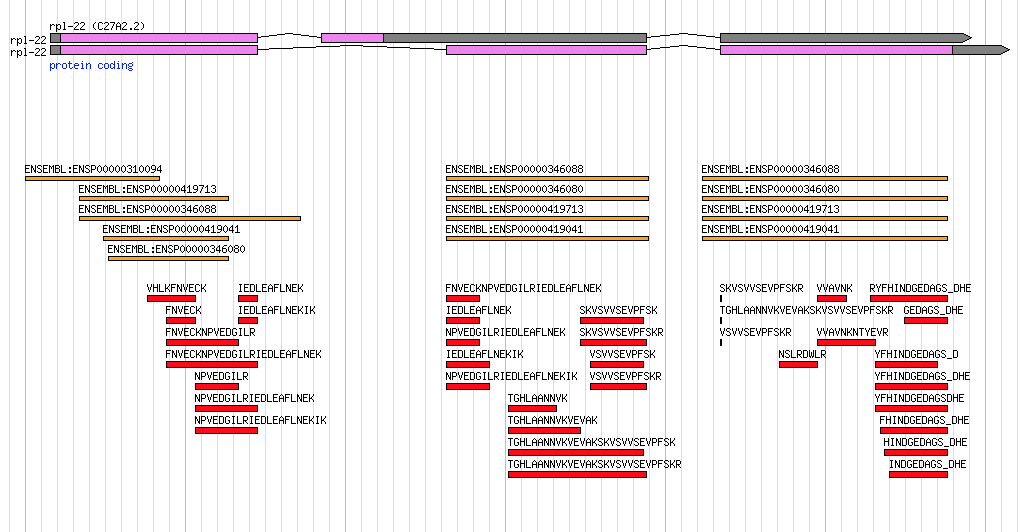
Indeed mass spec data has the potential to really clean up a lot of noisy gene annotations. In light of this I was very happy to see this new paper published in the Journal of Proteome Research (where talented up-and-coming scientists publish!):
Pooling data from 8 mass spec analyses of human data, the authors attempted to see how much protein support there was for the different annotated isoforms of the human genome. They could reliably map peptides to about two-thirds of the protein-coding genes from the GENCODE 20 gene set (Ensembl 76). What did they find?
We identified alternative splice isoforms for just 246 human genes; this clearly suggests that the vast majority of genes express a single main protein isoform.
They also found that the mass spec data was not always in agreement with the dominant isoforms that can be predicted from RNA-Seq data:
…part of the reason for this will be that more RNAseq reads map to longer sequences, it does suggest that either transcript expression is very different from protein expression for many genes or that transcript reconstruction methods may not be interpreting the RNAseq reads correctly.
The headline conclusion that mass spec evidence only supports alternate isoforms for 1.2% of human genes is thought provoking. It suggests to me that we should be careful in relying too heavily on gene annotations which describe large numbers of isoforms mostly on the basis of transcript data. Paraphrasing George Orwell:
All isoforms are equal, but some isoforms are more qual than others