
The three ages of CEGMA: thoughts on the slow burning popularity of some bioinformatics tools
The past
CEGMA is a bioinformatics tool that was originally developed to annotate a small set of genes in novel genome sequences that lack any annotation. The logic being that if you can at least annotate a small number of genes and have some confidence about their gene structure, you can then use them as a training set for an ab initio gene finder to go on and annotate the rest of the genome.
This tool was developed in 2005 and it took rejections from two different journals before the paper was finally published in 2007. We soon realized that the set of highly conserved eukaryotic genes that CEGMA used could also be adapted to assess the completeness of genome assemblies. Strictly speaking, we can use CEGMA to assess the completeness of the 'gene space' of genomes. Another publication followed in 2009, but CEGMA still didn't gain much attention.
CEGMA was then used as one of the assessment tools in the 2011 Assemblathon competition, and then again for the Assemblathon 2 contest. It's possible that these publications led to an explosion in the popularity of CEGMA. Alternatively, it may have become more popular just because more and more people have started to sequence genomes, and there is a growing need for tools to assess whether genome assemblies are any good.
The following graph shows the increase in citations to our two CEGMA papers since they were first published. I think it is unusual to see this sort of delayed growth in citations to a paper. The current citations from 2014 suggest that this year will see the citation count double compared to 2013.

The present
All of this is both good and bad. It is always good to see a bioinformatics tool actively being used, and it is always nice to have your papers cited. However, it's bad because the principal developer left our group many years ago and I have been left to support CEGMA without sufficient time or resources to do so. I will be honest and say that it can be a real pain to even get CEGMA installed (especially on some flavors of Linux). You need to separately install NCBI BLAST+, HMMER, geneid, and genewise, and you can't just use any version of these tools either. These installation problems have meant that I recently tried making it easier for people to submit jobs to us, which I run locally on their behalf.
These submission requests made me realize that many people are using CEGMA to assess the quality of transcriptomes as well as genomes. This is not something we ever advocated, but it seems to work. These submissions have also let me take a look at whether CEGMA is performing as expected with respect to the N50 lengths of the genomes/transcriptomes being assessed (I can't use NG50 which I would prefer, because I don't know the expected size for these genomes).
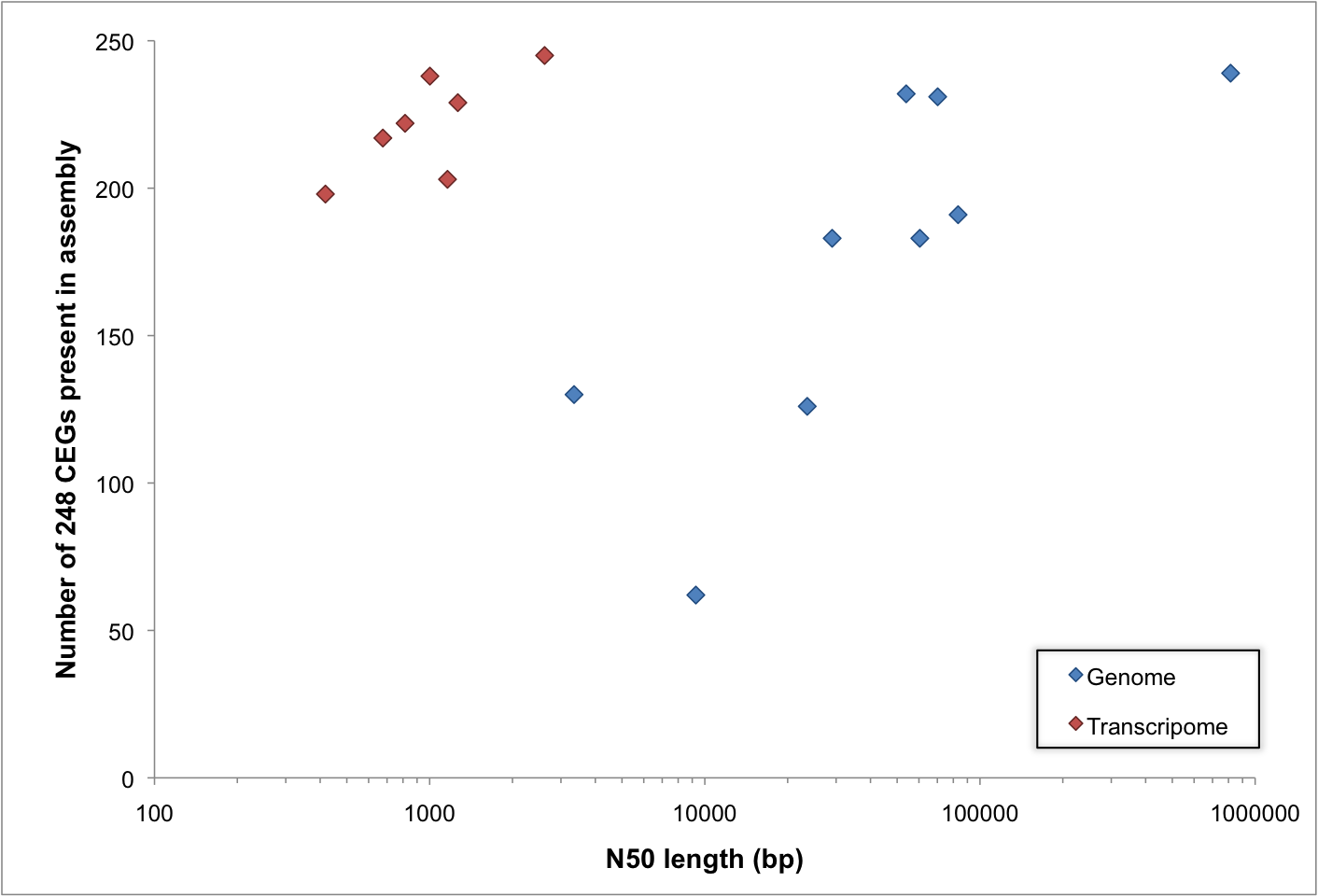
Generally speaking, if your genome contains longer sequences, then you have more chance of those sequences containing some of the 248 core genes that CEGMA searches for. This is not exactly rocket science, but I still find it surprising — not to mention worrying — that there are a lot of extremely low quality genome assemblies out there, which might not be useful for anything.
The future
We are currently preparing a grant that would hopefully allow us to give CEGMA a much needed overhaul. Currently we have insufficient resources to really support the current version of CEGMA, but we have many good ideas for how we could improve it. Most notably, we would want to make new sets of core genes based on modern resources such as the eggNOG database. The core genes that CEGMA uses were determined from an analysis of the KOGs database which is now over a decade old! A lot has changed in the world of genomics since then.











